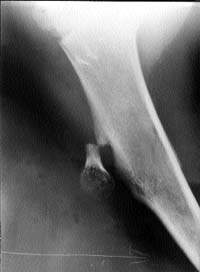
Рисунок 1. Большой экзостоз на ножке на границе верхней и средней третей плечевой кости. Отлом после случайной детской травмы
Наследственные полипозы желудочно-кишечного тракта (ЖКТ), особенно у детей, принято считать редкой патологией. Однако любая редкая патология перестает быть редкой, как только ее начинают целенаправленно изучать. Длительность интервала между появлением патологии и ее манифестацией определяется только компенсаторными возможностями (обратная пропорциональность с пенетрантностью гена), профилактическими мероприятиями и информативностью применяемых методик диагностики.
Наследственные полипозы заложены уже в генах родителей, а проявляются (боли в животе, кровавый стул, малигнизация) в массе своей на второй-третьей и даже четвертой декадах жизни и позже, хотя постепенно формируются уже у детей. Существует, однако, ряд внешних признаков (гипермобильность суставов, гамартомы, остеомы, пигментные пятна), которые по закону сцепленных генов с высокой долей вероятности позволяют высказаться в пользу системной наследственной патологии. Между тем даже эндоскопические находки полипозов ЖКТ редко заставляют врачей интерпретировать их как местное проявление общего состояния организма.
В связи с этим мы решили осветить проблему полипозов ЖКТ как системную динамическую патологию.
Существует большое число классификаций полипозов ЖКТ и их таксономической трактовки [7], что свидетельствует о сочетании этой патологии с другими наследственными состояниями, а кожные проявления сближают описываемую группу синдромов с факоматозами [9]. Приводим описание некоторых вариантов полипозов.
Бандлера синдром — гемангиоматоз тонкой кишки с пятнистой пигментацией кожи и слизистых. Существует мнение [11], что данное состояние имеет связь с синдромами Пейтц-Егерса и голубых сосудистых невусов (blue-rubber-bleb-naevi-syndrome). Клинически отмечается грубая пигментация по типу эфелид, пятен цвета “кофе с молоком” с преимущественной локализацией на лице, коньюнктиве, слизистой губ и рта. Манифестирует иногда уже с рождения ребенка. Боли в животе сочетаются с кровотечениями и развитием вторичной железодефицитной анемии. Синдром Бандлера рассматривают и в соотношении с болезнью Рендю-Ослера.
Бина синдром — вариант синдрома голубых невусов. Особым образом сформированные множественные кавернозные висцеральные и кожные гемангиомы. Гистологически они выглядят своеобразно: гемангиоматоз или гамартоз с полипозной структурой. Могут возникать всюду, в т. ч. на подошвах, что характеризуется кровотечениями вследствие травматизации [10]. Ангиомы кожи выглядят как синюшно-фиолетовые узлы (“винные пятна”), мягкие и безболезненные. Наблюдаются множественные кровоточащие гемангиомы в тонкой кишке (оккультные кишечные кровотечения), плевре, легких, печени, почках, скелетной мускулатуре. Типична вторичная постгеморрагическая анемия. Для глубоко расположенных гемангиом характерен положительный потовый тест Мануила-Ядассона: половина тела, где расположены ангиомы, потеет сильнее. Ангиомы выявляются эхографически, КТ, в момент оперативного вмешательства на кишечнике — при просвечивании кишечника эндоскопом. При этом на фоне ровного матового цвета стенки кишки обнаруживается вишнево-красный узел. В одном нашем наблюдении и в наблюдении M. Frosch [10] синдром сочетался с признаками долихостеномелии. В другом нашем случае у пробанда-девочки была долихостеномелия, а у ее старшей сестры наблюдалось преждевременное половое созревание и депигментированные пятна.
Дифференциальная диагностика проводится, наряду с полипозами ЖКТ, со всеми типами ангиоматозов.
Гарднера синдром — аутосомно/доминантно (а/д) наследуемый полипоз ЖКТ, особенно толстой кишки, сочетающийся с остеомами и опухолями мягких тканей. Около 50% всех случаев — спорадические. Проявляется ворсинчатым и аденоматозным полипозом вначале в толстой кишке, а затем и в желудке (неопределенные боли в животе, кровотечения), множественными остеомами плоских костей черепа (facies leonidica), аномалиями зубов, остеомами ребер, длинных трубчатых костей. На коже обнаруживают множественные дермоидные кисты, кисты сальных желез, фибромы, лейомиомы, пигментные пятна с обызвествлением последних. Наряду с этим описаны акроэритема, эритема над коленями, локтями, лодыжками, гиперкератоз ладоней и подошв, изменения ногтей по типу “часовых стекол” и их аномалии, перианальный зуд, диффузное облысение. Развиваются интра-, экстрамезентериальный фиброматоз, бронхоэктазы. Характерно также фиброматозное разрастание в области послеоперационных рубцов с развитием стриктур. Часто (до 12%) возникают опухоли головки поджелудочной железы, фатерова сосочка, надпочечников, щитовидной железы. Все вышеописанное сближает синдром со злокачественным аденоматозом (Гарднер II) и с семейным полипозом толстой кишки, при котором до 90% пациентов имеют остеомы нижней челюсти и/или аденомы 12-перстной кишки.
Олдфилда синдром — редкая комбинация семейного аденоматозного полипоза толстой кишки (см. ниже) с кистами сальных желез. В других случаях его рассматривают как вариант синдрома Гарднера. Передается а/д с различной степенью пенетрантности. Клинически протекает так же, как и семейный полипоз толстой кишки, но с образованием по всему телу воспаляющихся и рубцующихся кист сальных желез. В то время как кисты сальных желез образуются с рождения, полипы толстой кишки формируются только на втором-третьем десятилетиях жизни. Аденоматозные полипы часто малигнизируются, что резко ухудшает прогноз заболевания.
Пейтц-Егерса синдром — интестинальный полипоз типа II с пятнистой пигментацией кожи лица, слизистых губ и щек. Нельзя исключить, что он является вариантом ювенильного полипоза кишечника (см. ниже). Передается а/д с полной пенетрантностью, но различной экспрессивностью. При этом на коже лица, особенно вокруг рта, на слизистой рта, реже — над локтями, на подногтевых ложах появляются мелкие пигментные пятна цвета “кофе с молоком”, напоминающие веснушки. Пятна появляются в раннем детстве или имеются уже с рождения. В тонкой кишке, реже — в желудке и толстой кишке обнаруживаются множественные гамартомные полипы со сравнительно низкой вероятностью малигнизации. В 50% всех случаев полипы находят и у ближайших родственников.
Озлокачествление полипов наблюдается прежде всего при их манифестации уже в раннем детском возрасте.
Вероятность малигнизации возрастает после 35 лет [6]. Клиническая картина синдрома Пейтц-Егерса сводится к неспецифическим болям в животе, оккультным кровотечениям, железодефицитной анемии, хотя возможны илеус, синдром мальабсорбции с нарушением питания. В наших трех случаях синдром протекал на фоне долихостеномелии. В 5% случаев данный тип полипоза кишечника сочетается с гормонально активными опухолями яичников. Мы наблюдали сочетание синдрома Пейтц-Егерса с миксомным синдромом (опухолью сердца) у ребенка [5]. Этот же тип полипоза рассматривают и в соотношении с синдромами Реклингаузена и Бандлера (см. выше).
Наследственные полипозы — генетически детерминированные заболевания, клинически проявляющиеся чаще всего болями в животе и кровавым стулом во второй, третьей и даже четвертой декадах жизни
Полипоз генерализованный ювенильный — гастроинтестинальный полипоз (гамартомы и т. наз. ретенционные полипы — богатые соединительной тканью и покрытые слизеобразующим эпителием) с низкой вероятностью малигнизации. Передается а/д, хотя некоторые авторы описывают а/р (аутосомно-рецессивный) путь передачи летально протекающих вариантов.
Полипы встречаются чаще в небольшом числе, нередко — в прямой кишке. Уже в раннем детстве появляются спонтанные кишечные кровотечения, вторичная анемия, диарея с примесью слизи, иногда — инвагинация с непроходимостью. В 10% случаев полипы пролабируют трансанально или самостоятельно отходят. Возможны сочетания ювенильных полипозов кишечника с подкожными липомами, с артериовенозными аномалиями легких, кистами легких и почек. Хотя у детей этот тип полипоза протекает сравнительно благоприятно, с очень низкой вероятностью малигнизации, у ближайших родственников часто встречаются полипы толстой кишки и колонокарциномы.
Полипоз толстой кишки семейный — интестинальный полипоз типа 1, характеризуется распространенным диффузным полипозом толстой кишки. Начинается у детей (в 90% случаев полипы возникают в пубертатном периоде), но манифестирует в 20-30 лет. Почти в 2/3 случаев завершается мультицентрически растущей карциномой. Передается а/д с очень высокой пенетрантностью, так что не менее 50% ближайших родственников являются носителями полипов. Определена даже локализация гена — 5q (длинное плечо пятой хромосомы). Поэтому возможна генная диагностика носительства признака в асимптомных случаях. Фибробласты кожи как у пациентов, так и у бессимптомных членов семьи, задолго до клинической манифестации полипоза толстого кишечника, in vitro характеризуются нарушенным ростом и делением. Полипы — тубулярные, тубуло-ворсинчатые или ворсинчатые аденомы. Полипоз проявляется рецидивирующими поносами с кровью или, при локализации полипов в прямой кишке, со слизью. Вторично развивается железодефицитная анемия. В редких случаях полипы обнаруживают в желудке, 12-перстной кишке и терминальном отделе тонкой кишки — несмотря на то, что, в противоположность синдромам Гарднера и Тюрко, в этом случае не наблюдается сочетание с опухолями других мягких тканей. При семейном полипозе толстой кишки также рентгенологически обнаруживаются очаговые изменения костей (рис. 1). Прогноз семейного полипоза толстой кишки плохой. Через 10-15 лет после появления первых клинических признаков практически всегда обнаруживают мультицентрически растущую колонокарциному.
Тюрко синдром — редко встречающееся сочетание полипоза толстой и прямой кишки с опухолями мозга. Передается, вероятней всего, а/д. Около 40% случаев спорадические. Манифестирует симптомами семейного полипоза, а после полового созревания — симптомами опухолей мозга (глиобластома, медуллобластома).
Цанка синдром — то же самое, что и семейный полипоз кишечника (см. выше), но с множественными хрящевыми экзостозами на трубчатых костях и позвоночнике без поражения костей черепа. Передается а/д. Экзостозы формируются уже в раннем детском возрасте, но полипоз развивается во второй-третьей декадах жизни.
Истинная частота полипозов кишечника, особенно у детей, неизвестна. Часть больных попадает к нейрохирургам по поводу опухолей мозга, и в силу тяжести состояния им не проводится динамическая эндоскопия и рентгеноскопия желудочно-кишечного тракта с барием. Часть больных наблюдается в неврологических клиниках как пациенты с факоматозами.
Многочисленные соотношения полипозов кишечника с другими наследственными синдромами, с изменениями кожи, нервной ткани, иных систем, близость гистологических проявлений различных типов полипозов и временных этапов манифестации позволяют задать вопрос: всегда ли связь синдромов является случайной, или существует ряд переходных форм с разной экспрессивностью тех или иных сопутствующих признаков? Правомерно ли рассматривать эти формы как обособленные? Даже при точном генном анализе мы не получим скорого ответа, т. к. в одном и том же гене возможно множество мутаций [13]. При этом синдромы полипозов кишечника объединяются склонностью к опухолям (чаще множественным), пигментации кожи, гипермобильности суставов, гиперэластичности кожи, пролабированию клапанов сердца, нарушениям его ритма, выраженным нейро-эндокринным дисфункциям.
О полипозах кишечника следует думать при всех случаях нейрокутанных синдромов (пятна цвета “кофе с молоком”, ангиомы), фибромах, аденомах сальных желез, при синдромах гипермобильности, экзостозах, опухолях нервной ткани, мягких тканей и желез внутренней секреции, а тем более — при наличии полипов у родственника обследуемого.
Клиническая симптоматика полипозов кишечника маловыразительна и неспецифична. Вовсе не обязательно, чтобы в каждом случае присутствовали все вышеперечисленные для данного синдрома признаки: вариабельность их высока. Полипозы кишечника необходимо исключать при каждом случае кишечных кровотечений, рецидивирующих диспепсий, особенно с примесью крови или слизи.
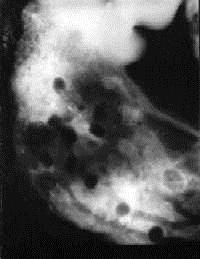
Рисунок 2. Рентгенограмма множественных полипозов желудка при синдроме Пейтц-Егерса, выглядящих как дефекты заполнения
Рентгенологически полипы выглядят как круглые дефекты наполнения с сохраненной структурой стенки кишки, складок и гаустраций (рис. 2). Гигантские полипы могут значительно затруднить продвижение химуса по кишечнику (рис. 3).
Схожая рентгенологическая картина наблюдается и при других состояниях, в частности, при псевдополипах, при возрастной гиперплазии лимфатических бляшек в стенке кишечника у детей [15] и при раке. Рентгенологически возвышенный тип рака имеет вид полиповидной опухоли, имеющей дольчатую или бугристую поверхность. Дефект наполнения имеет округлую или овальную формы с четкими контурами, “ободок” при использовании метода двойного контрастирования. Важно оценить основание опухоли, т. к. широкая ножка характерна для рака. В результате дегенеративных процессов центр опухоли западает, однако рентгенологически изъязвления определяются далеко не всегда. Все эти показатели не могут быть абсолютными — бывают случаи, когда полипы, диагностированные как злокачественные, при гистологическом исследовании оказываются доброкачественными [8].
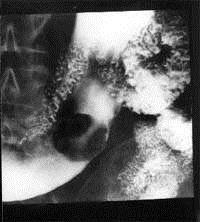
Рисунок 3. Рентгенограмма гигантского полипа на ножке в кишечнике
Эхографически полипы визуализируются при их диаметре не менее 8 мм и наличии ножки. Полипы видны как округлые образования на стенке кишки, выступающие в ее просвет. Наилучшим образом эхографически визуализируются полипы прямой кишки. При диффузном полипозе стенка кишки истончается, что не наблюдается при единичных полипах [4]. Но полноценная диагностика с применением гистологических методик возможна только по итогам эндоскопии.
Тактика врача. При всех впервые выявленных случаях полипозов должен быть проведен максимально возможный осмотр кишечника, т. е. рентгеноскопия тонкого кишечника с двойным контрастированием, эзофагогастродуоденоскопия и колоноскопия. Колоноскопия предпочтительнее сигмоскопии, т. к. при последней аденому не удается выявить в 30% случаев [6]. Только эндоскопия позволяет достоверно диагностировать полипоз и отличить его от карциномы, карциноида, эозинофильной гранулемы, лимфоидной гиперплазии слизистой оболочки, ювенильного полипа (киста, образующаяся при облитерации выводного протока железы желудка), болезни Менетрие, мелкоузловатой лимфосаркомы толстой кишки, аденом Бруннеровых желез, туберкулеза кишечника, псевдополипоза [1, 2, 3]. Биопсию, по крайне мере первую, желательно проводить в стационаре из-за опасности кровотечения. Одиночные полипы могут быть удалены эндоскопически. Регулярное проведение колоноскопий с интервалом в 6 месяцев с удалением полипов позволяет на несколько лет оттянуть выполнение тотальной колоноэктомии. При синдромах с высокой вероятностью малигнизации проводятся расширенные вмешательства вплоть до колоноэктомии.
При обнаружении полипов кишечника и получении результатов гистологического исследования необходимо немедленно обследовать родственников больного. Причем не только с точки зрения наличия возможного полипоза, но и для выявления иной патологии (опухолей мозга и т. д.). При семейных аденоматозных полипозах (синдромы Гарднера, Тюрко и др.) колоноскопию с полипэктомией необходимо проводить не реже одного раза в 6 месяцев. У родственников первой степени родства ректосигмоскопию надо проводить не реже одного раза в год. При двух подряд следующих отрицательных результатах, отсутствии кожных пятен и клинической симптоматики частоту эндоскопий можно снизить до одной в 2-3 года. При обнаружении аденом необходима полная колоноскопия. Поскольку семейный аденоматозный полипоз представляет собой облигатное предраковое состояние, Классен [6] рекомендует превентивную колоноэктомию. При наложении илеоректального анастомоза эндоскопический контроль в послеоперационном периоде проводят раз в 4-6 месяцев, при илеоанальном анастомозе — раз в 1 год. Раз в 1-3 года проводят эзофагогастродуоденоскопию с обязательным удалением возможных полипов и аденом 12-перстной кишки. У членов семьи, у которых ген 5q (ген семейного аденоматозного полипоза) не обнаружен, колоноскопию можно проводить с интервалом 3-5 лет [6].
При всех впервые выявленных случаях полипозов необходимо провести максимально полное обследование кишечника, включающее рентгеноскопию тонкого кишечника с двойным контрастированием, эзофагодуоденогастроскопию и колоноскопию
При синдроме Пейтц-Егерса риск малигнизации прямо пропорционален возрасту больного. Поэтому пациенты старше 35 лет должны регулярно проходить эндоскопический контроль верхних и нижних отделов ЖКТ, а также рентгенологическое исследование тонкой кишки с двойным контрастированием. При полипах с низким риском малигнизации или не склонных к озлокачествлению, расположенных в толстой кишке, проводят их удаление через эндоскоп. Причем можно подождать, пока у полипа сформируется ножка, — в этом случае его удаление становится менее рискованным. Место бывшей локализации полипа метят китайской тушью для обеспечения точного топического наблюдения. Такая тактика оправданна очень слабо выраженной сетью лимфатических сосудов, что отодвигает срок возможного метастазирования. Полипы, расположенные в желудке и тонкой кишке, т. е. там, где лимфатические сосуды развиты, требуют значительно более активной тактики лечения и расширенного вмешательства [12, 16, 17].
Больным с полипозами и их родственникам необходимо избегать вероятных канцерогенных факторов: токи высокой частоты, витамины Д2, В12, фототерапия и др.
Таким образом, мы должны помнить о возможном полипозе ЖКТ при многих системных заболеваниях. В свою очередь, обнаружение полипов требует тщательного и целенаправленного клинико-инструментального системного обследования пациента, его родственников, динамического контроля, онкологической настороженности врача. Полноценный врачебный подход к проблеме, объединение усилий терапевта, педиатра, эндоскопистов, гистологов, генетиков и других специалистов, принцип “единоначалия” в ведении таких больных — все это способно значительно изменить наши представления о семейных полипозах, их таксономическом месте и генетически определенных предраковых синдромах.